CRISPRclean®
1 /3Pages

CRISPRclean®
1 /3Pages
Catalog excerpts

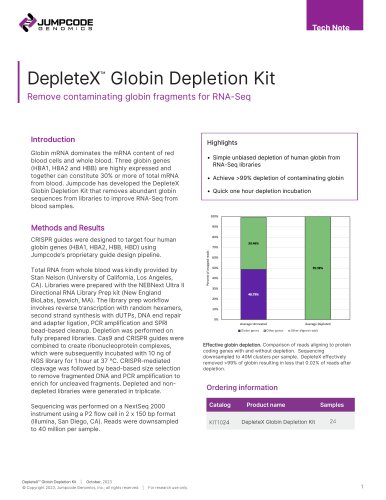
TECH NOTE CRISPRclean® depletion technology: A new tool to better understand rare diseases Enhanced RNA-Seq delivers double the genes detected with confidence Introduction Current first-line genetic tests for rare diseases include chromosomal microarray (CMA) and clinical exome sequencing. However, these methods are insufficient for a large proportion of cases. Whole genome sequencing (WGS) is commonly used to detect rare structural variants and small copy number variations (CNVs). Combining RNA sequencing (RNA-Seq) with WGS promises to enhance our understanding of rare diseases’ mechanisms.1-2 This approach allows researchers to observe the aberrant transcription of RNA from DNA by allowing the calculation of the splicing ratio for novel unannotated junctions and reporting the proportion of reads that support aberrant splicing. Significantly, RNA-Seq increases the detection of low-expressing transcripts and isoforms for rare diseases.3-7 Typically, most genes in Online Mendelian Inheritance in Man (OMIM) are expressed in accessible tissues from any given patient, though at low levels and cannot be used for splicing analysis. Many RNA-Seq reads originate from abundantly expressed genes and are irrelevant to a given rare disease etiology. However, it is possible to remove this abundant noise before sequencing by targeting genes with transcripts per million (TPM) > 30. Removing these high-expression genes results in sequencing reads being redistributed to low and medium-expressed genes, providing a more confident sampling of this minimally observed part of the transcriptome. The increased coverage significantly boosts the detection sensitivity and confidence of aberrant gene expression signals. Here we show that using CRISPRclean technology, abundant and uninformative RNAs can be easily removed from NGS libraries. This approach leverages the in vitro depletion of library fragments with CRISPR/Cas9 complexes CRISPRclean® High Expressing RNA Depletion Kit © Copyright 2022, Jumpcode Genomics, Inc.; all rights reserved programmed with guide RNAs to desired targets to be removed. The application of CRISPRclean technology to fibroblast samples doubles the number of observed genes at TPM > 30 because of increased coverage from reassigned reads, thereby increasing the reliability of analysis and expanding discovery potential. Methods Three fibroblast total RNA samples were split into two aliquots and depleted in triplicate using ~450,000 guides designed against 4,326 high-expression genes (> 30 TPM). Guides were designed to target 70% of the total raw reads for depletion. Control (non-depleted) samples were retained in triplicate from the second aliquot. Sequencing libraries were generated from control, and post-depletion replicates using the NEBNext® Ultra II Stranded Total RNA with Poly A module kit. Size selection of the final library was performed at 400-500 bases, and libraries were sequenced on the NextSeq® 2000 as 2x150 base reads. All samples were normalized to 100 MM reads for analysis. Results Depletion significantly increases the coverage of low and medium-expression genes The sequencing results of the fibroblast samples show a significant boost in coverage for low and medium-expression genes for the depleted samples when compared to the control. In Figure 1, the purple population of genes is the low to medium-expression genes in the CRISPRclean depleted condition, which is now covered at TPM > 30. These genes are not covered at that level in the control sample. Genes with TPM
Open the catalog to page 1
TECH NOTE 30 in both the control and depleted samples are shown in blue. 4,477 genes were raised above a threshold of 30 TPM due to depletion in the CRISPRclean treated sample. This represents 4,477 genes that now have sufficient coverage for rare disease researchers to proceed with further analysis reliably. TPM of greater than 30 in the control to the depleted sample, there are 3380 genes in the control population and 6425 genes in the depleted sample. Thus, using CRISPRclean depletion to boost low and mediumexpression genes doubles the discovery space for the fibroblast samples. DEPL (6425)...
Open the catalog to page 2
TECH NOTE Ordering information Catalog Product name CRISPRclean® High Expressing RNA Depletion Kit Cummings, B. B., Marshall, J. L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A. R., Bolduc, V., Waddell, L. B., Sandaradura, S. A., O’Grady, G. L., Estrella, E., Reddy, H. M., Zhao, F., Weisburd, B., Karczewski, K. J., O’Donnell-Luria, A. H., Birnbaum, D., Sarkozy, A., Hu, Y., Gonorazky, H., … MacArthur, D. G. (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Science translational medicine, 9(386), eaal5209. https://doi.org/10.1126/scitranslmed.aal5209...
Open the catalog to page 3All Jumpcode catalogs and technical brochures
 DepleteX® Mito DNA Depletion Kit
DepleteX® Mito DNA Depletion Kit11 Pages